There are plenty of medical devices, beginning from simple and non-risky medical devices and ending in invasive implants and combination products combined with a drug product delivery system.
It is a long journey to develop a new medical device until it is approved in the target markets such as the FDA, Israel, and CE mark. This journey begins in the design phase and requires patience, budget, and professionalism.
There are many challenges along the way to international success.
Medical devices’ safety, functionality, and claims must be proven, unlike other products. The medical device must demonstrate safety and efficacy as part of clinical studies and evaluation before the medical device is finally registered in the relevant health authorities.
Medical Device Market Opportunity Study
For the developers and manufacturers of medical devices, there must be a viable population of candidates/patients to benefit from their medical devices, solutions, and services.
Before you start, the potential beneficiaries and opportunity market should be clearly identified and as early as possible. The market’s size will inform industry economic and behavioral decisions. If the target market is too small or the business model is wrong, the company may not find product viability.
A large market can support a public company, whereas smaller markets may not. Many small markets can still support a healthy private company, which could be an attractive acquisition target. Market size also affects the organizational structure, like how easy it will be to raise capital.
There are established markets where a new product can take a percentage of competing alternatives’ market share. There are also unestablished areas where a novel medical device can be utilized without current alternatives. However, before a comprehensive review of potential market opportunities can begin, all factors should be considered.
As part of the business model and financial calculations, there are multiple factors that should be analyzed, for example:
- Recurrent device use
- Implementation
- Market data reliability
- Marketing strategy
- Economic changes
- Political changes
- Regulatory strategy
- Governmental regulations
Medical Device Proof of Concept
The purpose of the proof of concept stage is to demonstrate that the idea can actually work. It is a pre-development stage activity and is therefore excluded from regulatory frameworks.
Proofs are done when there are new applications of an existing engineering or scientific principle, demonstrating that critical design features of the medical device concept perform as intended.
Early device prototypes address design challenges. When more than one critical process is addressed, they will need their own proof. For example, a software-inclusive implant may need software and hardware proof of concepts.
While not business-oriented, they impact potential industry activity by informing stakeholders involved in the medical device risk analysis and usability assessment to look at potential failures and hazards comprehensively. Proof of concept also helps mitigate design risks, if any, before getting into the full product design mode.
The next step is to show that the medical device can be manufactured by creating a prototype and applying manufacturing engineering principles. Then development starts based on that initial medical device design attempt and when the decision to make/market a device is made, along with the usual beginning development phase activity.
Medical Device POC Activities examples:
- Medical device prototype critical design features
- Medical device prototype critical manufacturing stages
- Develop preliminary medical device test methods
- Procure or design preliminary test equipment
- Perform focus groups if required
- Conduct design reviews as required
- Initiate the development of a risk management plan
- Determine medical device regulatory strategy
- Define medical device packaging and labeling requirements
- Define medical device sterility method and bioburden requirements
Medical Device POC Deliverables examples:
- Proven concepts
- Completed Design Requirements Document (DRD)
- Prototypes of critical design areas or processes
- Preliminary test methods
- A draft risk management plan
- A draft risk assessment
Medical Device Design Specifications
A crucial part of the medical device design and development process is to create a product requirement specification.
The document takes place in the very earliest stages of the process. It ensures the design engineers understand what is expected and how the product’s success will be measured.
Medical device design requirement specification also allows you to demonstrate the product does what it was designed to do (claim). It can also help with the regulatory compliance process, as it helps you demonstrate the design process was conducted correctly.
A requirement is a statement of one thing a product must do or a quality/safety feature it must have.
A requirement specification is a collection of the set of all requirements to be imposed on the design and verification of the product and other related information necessary for the design, verification, validation, and maintenance of the product.
Medical Device Prototyping and Product Development
Prototyping is important to prove device safety, functionality, cost-effectiveness, and regulatory feasibility.
This early stage allows medical engineering teams to understand the device’s purposes and identify areas for design improvement.
The creation of the first medical device prototype is based on initial detailed design drawings. This prototype is a crucial part of the iterative design process and is mainly used to improve and enhance the design.
Unlike a decade ago, nowadays, a medical device prototype can be made using small scall 3D printing devices relatively easily.
Medical Device Alpha Prototype
The medical device Alpha prototype is the “initial attempt” at designing and fabricating the product to meet the Product Requirements Specification (PRS).
It is the first attempt to make a medical device prototype that both looks like and works like the final product.
The iterative process of designing and building the Alpha prototype will provide guidance for the next stage. It will have testing and evaluation of performance, safety, EMC, usability, and appearance.
Alpha prototype of medical device development is typically the most expensive stage, requiring months of reiteration and refinement.
Alpha medical device design and testing are essential to understanding the device’s limitations and objectives in refining the design.
This medical device development phase will involve design specifications (DS) for hardware and software that define performance specifications and incorporate safety mitigation.
At the end of the Alpha prototype development stage, device requirements, risk management, regulatory strategy, verification & validation (V&V) plans, and other regulatory requirements and documents, should be defined towards the FDA pre-submission meeting.
The pre-submission meeting is not mandatory. It can benefit the company by receiving FDA inputs and, in theory, usually will clarify the selected regulatory pathway and the required testing plan.
It is best practice to receive FDA feedback before embarking on Beta prototype development.
Medical Device Beta Prototype
The medical device Beta prototype development incorporates the design refinements in Alpha development and implements them into production modeling.
Test plans and verification protocols are prepared at this stage. The software is refined and ready for the first release.
Documentation is updated and prepared for releasing the device master record (DMR), and Medical Device production testing and assembly protocols are drafted, Beta prototypes are assembled and tested as aligned with the production procedures, and hazard mitigations are documented in a risk assessment report. The Beta prototypes are ready for verification and preliminary validation testing, safety and EMC testing, and performance testing to verify compliance with the PRS.
Medical device prototype refinements will be required after the assembly of the Beta prototypes. These should be under configuration control to reflect the reasons for the changes and how they make the Beta prototype overcome any deficiencies in meeting specifications and standards.
Medical Device Beta prototype development will involve the development of verification specifications for hardware and software to ensure that the medical device meets design requirements.
Medical Device Pilot Prototype
The medical device pilot production phase is where the Beta prototype verification and validation testing refinements are incorporated into the design and production process.
The documentation for the Design Master Record (DMR) is updated, and the design of manufacturing and the implementation of the quality management system (QMS) is done for pilot production.
These medical device units may be used for usability testing, different studies, and clinical trials and are prerequisites before initial release to market.
The medical device design and the production process are far more streamlined. Submission to the FDA and/or CE mark for regulatory release to market is completed during this phase.
Final Medical device
The final medical device incorporates the refinements from usability studies and production monitoring.
The design and the medical device assembly process are stable, repeatable, have high yields, and incorporate cost-saving measures, and are fully validated.
The medical device’s post-market surveillance (PMS) must be implemented, collecting feedback from complaints, user requests, and production data.
This feedback may result in initiating a Corrective and Preventative Action Plan (CAPA) to resolve any issues.
The creation of the final medical device prototype is the end of the design process.
A small batch of the prototype will be manufactured to be used for testing and evaluation. This prototype must be validated and regulatorily compliant.
Medical Device Design Controls and cGMP
The purpose of the design control process is to document and control the medical device design process, to ensure that devices meet all requirements specifications and user needs such as intended use, risks, and safety requirements.
As part of the design control, special attention should be given to:
- Medical device design and development planning
- Identifying medical device design inputs
- Developing medical device design outputs
- Verifying that medical device design outputs meet design inputs
- Validating the medical device design
- Controlling medical device design changes
- Reviewing medical device design results
- Transferring the medical device design to production
- Compiling a design history file (DHF) to ensure that resulting designs will meet user needs, intended uses, and requirements.
All medical device manufacturers in the U.S. must comply with the FDA’s Quality System Regulations (QSR). Also known as the Good Manufacturing Practice (cGMP) requirements and ISO 13485.
The QSR was updated in 1990 to include design controls authorized by the Safe Medical Devices Act.
To create consistent requirements for quality systems, the FDA opted to update the International Organization for Standards (ISO) 9001:1994 “Quality Systems–Model for Quality Assurance in Design, Development, Production, Installation, and Servicing,” eventually resulting in the ISO/CD 13485 “Quality Systems–Medical Devices–Supplementary Requirements to ISO 9001.”
Because of the broad spectrum of the medical device industry, it’s not practical for the FDA to prescribe how manufacturers should produce medical devices. Instead, the QSR serves as a framework for all manufacturers to follow.
Still, medical device manufacturers are tasked with following appropriate best practices, establishing suitable medical device requirements for safety and efficacy, and developing methods and procedures for designing, producing, and distributing devices in line with the quality system requirements. In other words, the QSR outlines the end goal at a “high level” but leaves the interpretation for creating the road map for getting there up to manufacturers. Of course, these interpretations will be examined as part of the regulatory audit afterward.
Design controls are the policies, processes, and procedures for managing design activities, evaluating quality, and adjusting for errors and shortcomings throughout the medical device development process.
Medical Device manufacturers must have their quality systems in place – defined and implemented – before they go to market.
Most medical device startups create their quality systems as they go, but all parts must be fully implemented correctly before regulatory submission.
Medical Device QMS [ISO 13485 and 21 CFR part 820]
ISO 13485:2016 specifies requirements for a quality management system (QMS). Organizations must demonstrate the ability to provide medical devices and related services that consistently meet customer and regulatory requirements. Such organizations can be involved in one or more parts of the development process, including design and development, production, storage and distribution, post-marketing surveillance, etc.
The requirements of the ISO 13485:2016 apply to organizations regardless of their size and type except where explicitly stated. Wherever requirements are specified as applying to medical devices, the requirements apply equally to associated services as supplied by the organization.
The processes required by ISO 13485:2016 that apply to the organization, but are not performed by the organization, are the responsibility of the organization and are accounted for in the organization’s quality management system by monitoring, maintaining, and controlling the processes. Third-party auditors will inspect every quality management aspect carefully and therefore, it is recommended to use QMS consultancy accordingly.
If applicable regulatory requirements permit exclusions of design and development controls, this can be used as a justification for their exclusion from the quality management system. These regulatory requirements can provide alternative approaches to be addressed in the quality management system.
It is the responsibility of the organization to ensure that claims of conformity to ISO 13485:2016 reflect any exclusion of design and development controls.
21 CFR Part 820 is part of the Current Good Manufacturing Practice (cGMP) regulations. It proves that devices are built under a quality system. It outlines quality systems requirements along with a slew of others, including design, document, and purchasing controls, among twelve other subheadings.
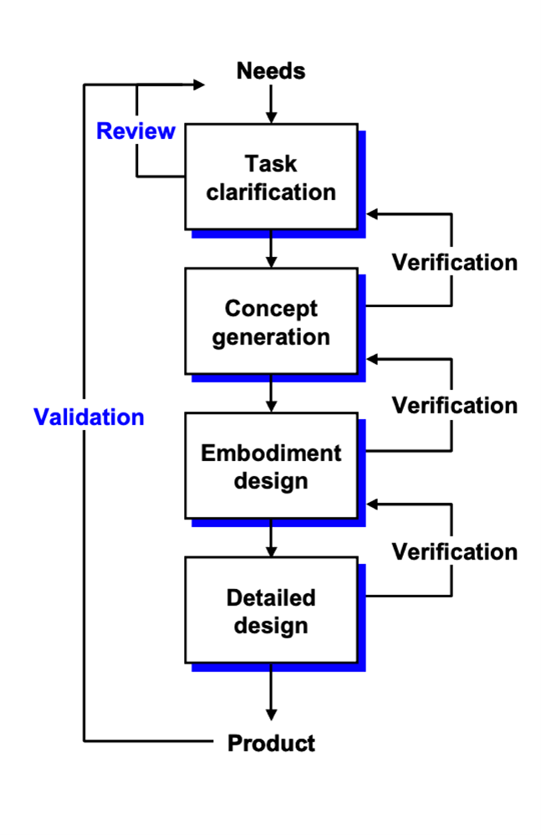
Medical Device Verification and Validation (V&V)
Verification and validation (V&V) are checks and balances in the medical device design process that identify deficiencies and discrepancies in the design before the device is produced.
Verification is particularly important in medical device design, while the validation is before the industrial manufacturing phase.
Verification ensures the output of each phase of the design process meets the requirements derived from the previous phase. Verification activities may include:
- Worst-case analysis
- Fault tree analysis (FTA)
- Inspection
- Testing
- Failure mode and effects analysis (FMEA)
Medical device verification tests the output from each step against the previous step.
Validation ensures that the product meets the user’s needs and prioritizes risks. In medical device design, verification and validation must be done to obtain approval for the device.
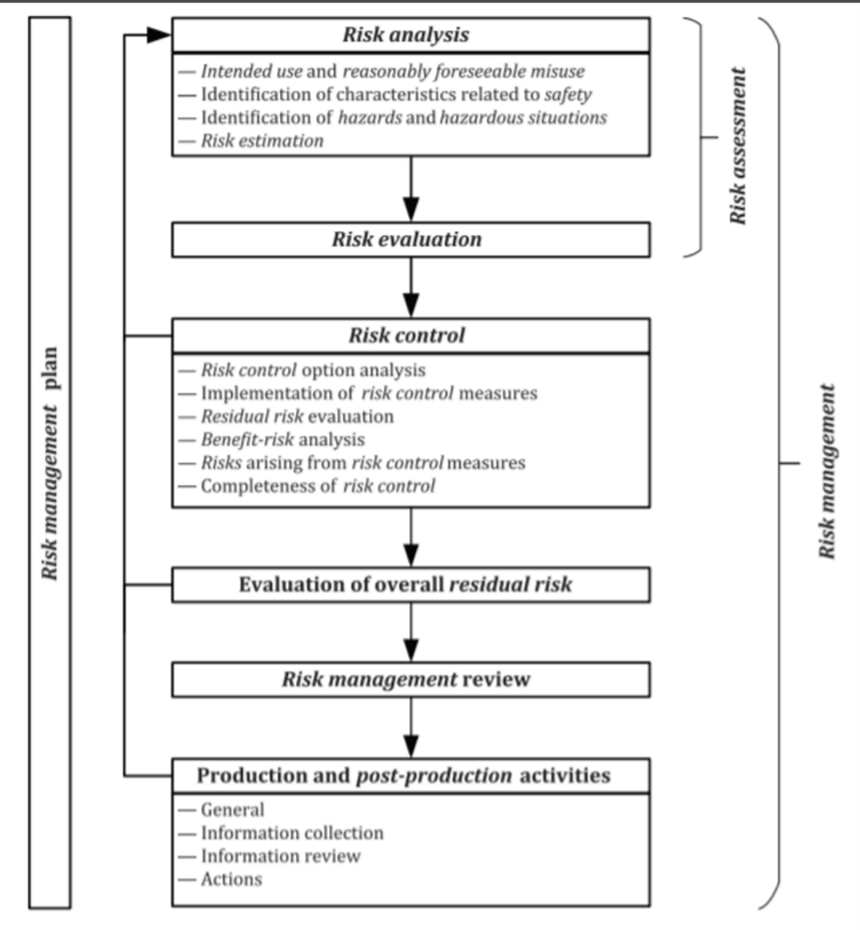
Medical Device Risk Management and Risk Assessment
The FDA routinely emphasizes a “risk-based approach,” and medical device production is included.
In simple terms, the FDA wants to maximize benefits while minimizing harm and hazards.
This falls under Risk Management, a systemic application of management policies, procedures, plans, and practices to analyze, evaluate, control, and monitor risks.
A significant aspect of this mission includes a risk assessment with both risk analysis and a risk evaluation related to the medical device.
Risk analysis is the systematic use of available information to identify hazards and estimate the risk. Risk evaluation is the process of comparing the calculated risk against given risk criteria to determine the acceptability of the risk.
The FDA has a diagram to visualize risk procedure processes through the production lifecycle. From the initial risk analysis of the concept to post-production activities, there are constant safety checks to protect patient safety.
More information on the extensive risk-based protocol can be found in the diagram below:
Risk Prioritization in Failure Mode and Effects Analysis (FMEA)
The purpose of the FMEA process is to identify, analyze, and evaluate risks, often associated with the development and production of a medical device.
Examples of risks assessed by FMEA are:
- Non-quality product risk and its impact on patient health
- Risk analysis of medical software, computerized system, or application
- Risk analysis of production infrastructure
- Risk analysis of production system or equipment
- Risk of nonconformities or deviations,
- Analysis of risks that affect the safety of users of the product
- Analysis of risks after changes and improvements
The goal of risk analysis in the FMEA methodology is to document the potential risks in systematic analysis and to quantify and rank the risks to define priorities for corrective and preventative actions if required (depending on the level of risk).
The FMEA process should be conducted in conjunction with the most heterogeneous professional teams (engineering, quality, safety, production, procurement, logistics, etc.). As much data as possible should be gathered prior to the risk analysis process to be a factual basis for decision-making.
The risk priority number (RPN) will be determined based on different parameters. Risk should usually be addressed at medium and high rating levels.
The identified prioritized risks will be mitigated by defining corrective actions, preventive actions, and the addition of appropriate and effective risk controls.
The main products of the medical device FMEA process are:
- Identification of risks and failures
- Reduction in the severity of the result of a failure
- A reduction in the probability of failure
- Improve the ability to identify a failure in a timely manner
- Treatment of failures and risks in relevant rankings only
- Defining corrective and preventive actions while monitoring and evaluating the effectiveness
Medical Device classification
The FDA categorizes medical devices into one of three classes – Class I, II, or III – based on their risks and the regulatory controls necessary to ensure safety and effectiveness.
Medical Class I devices generally pose the lowest risk to the patient and/or user, and Class III devices pose the highest risk. Device classification also depends on the intended uses, invasiveness, and indications for use.
The class to which your medical device is assigned determines, among other things, the type of premarketing submission/application required for FDA clearance to market.
If your device is classified as Class I or II, and if it is not exempt, a 510k will be required for marketing.
All medical devices classified as exempt are subject to the limitations on exemptions covered under 21 CFR 862-892.
A premarket approval application (PMA) for Class III medical devices will be required.
Medical Device Testing Strategies and Performance Parameters
Medical device testing is the process of demonstrating that the device will reliably and safely perform in use.
In new medical device development, extensive Design Validation Testing is applied. This includes performance testing, biocompatibility, toxicity, chemical analysis, and sometimes human factors or even clinical testing.
Ongoing quality assurance and quality control testing are generally more limited. This usually includes dimensional checks, functional tests, and packaging verification.
Material characterization has many applications in medical device development and production. It is the chemical analysis of materials to identify chemical composition. This information is then used to minimize toxicological testing of new designs as well as for production and design changes.
Medical devices require varying degrees of biocompatibility testing according to their classification. The main source of guidance on the essential requirements for biological safety is ISO 10993 – Biological Evaluation of Medical Devices. This standard defines devices in terms of their invasiveness and the duration of patient contact, which subsequently determines what level of safety testing manufacturers must complete before putting their devices on the market.
Each type of medical device, from software to contact lenses to pacemakers, will have testing strategies associated with their unique type to identify hazards in clinical use.
Non-clinical bench performance testing can be completed by either the medical device manufacturer or a third-party testing facility, depending on the device or device type. It covers mechanical and biological engineering performance such as fatigue, wear, tensile strength, compression, and burst pressure. Bench tests using ex vivo, in vitro, and in situ animal or human tissue; and animal carcass or human cadaver testing, FDA said.
The testing excludes biocompatibility evaluation, reprocessing or sterilization validation, human factors, software verification and validation, and computational modeling.
Test reports for clinical studies, animal studies, and additional studies evaluating the performance characteristics of in vitro diagnostic devices.
Medical Device performance testing requirements can be found below:
- IEC/AAMI 60601, Medical electrical equipment – General requirements for basic safety and performance
- IEC 60601-1-2, Electromagnetic compatibility (EMC), wireless testing, coexistence
- IEC 61010, Safety requirements for electrical equipment for measurement, control, and laboratory use
- IEC 61326-1, Electrical equipment for measurement, control and laboratory use – EMC requirements – Part 1: General requirements
- IEC 61326-2-6, Electrical equipment for measurement, control, and laboratory use – EMC requirements – Part 2-6: Particular requirements – In-vitro diagnostic (IVD) medical equipment
- UL 1069, the Standard for hospital signaling and nurse call equipment
- UL 2560, the Standard for emergency call systems for assisted living and independent living facilities
- UL 1431, the Standard for personal hygiene and healthcare appliances
- IEC 62304, the Standard for medical device software
- IEC 62366-1, the Standard for the application of usability engineering to medical devices
- ANSI/CAN/UL 2900-1, the Standard for software cybersecurity for network-connectable products, Part 1: General requirements
- ANSI/CAN/UL 2900-2-1, the Standard for software cybersecurity for network-connectable products, Part 2-1: Particular requirements for network-connectable components of healthcare and wellness systems
Gain the advantage of bringing your Medical Device to the global market by comprehending the developmental and regulatory journey.
This knowledge will empower you to secure approvals, complete registrations, and achieve international sales, including in regulated markets like the US, EU, Japan, Australia, Canada, and Israel.
Take the first step towards your success now!