ML and AI technologies for software as a medical device
AI (Artificial Intelligence) is shorthand for any task a computer can perform in a way that equals or surpasses human capability.
It makes use of varied methods such as knowledge bases, expert systems, and machine learning. Using computer algorithms, AI can sift through large amounts of raw data looking for patterns and connections much more efficiently, accurately, and quickly than a human could.
Artificial intelligence and machine learning technologies have the potential to transform health care by deriving new and important insights from the vast amount of data generated during the delivery of health care every day.
Medical device manufacturers, as well as medical software developers, are using these technologies to innovate their products and/or services to assist healthcare providers better and improve patient care.
One of the greatest benefits of AI/ML in medical software resides in its ability to learn from real-world use and experience and its capability to improve its performance over time.
SaMD regulations in the FDA
The FDA’s traditional paradigm of medical device regulation was not designed for adaptive artificial intelligence and machine learning technologies.
On April 2, 2019, the FDA published a discussion paper that describes the FDA’s foundation for a potential approach to premarket review for artificial intelligence and machine learning-driven medical software. The ideas described in the discussion paper leverage practices from our current premarket programs and rely on IMDRF’s risk categorization principles, the FDA’s benefit-risk framework, risk management principles described in the software modifications guidance, and the organization-based total product lifecycle approach.
In the framework described in the discussion paper, the FDA envisions a “predetermined change control plan” in premarket submissions. This plan would include the types of anticipated modifications—referred to as the “Software as a Medical Device Pre-Specifications”—and the associated methodology being used to implement those changes in a controlled manner that manages risks to patients —referred to as the “Algorithm Change Protocol.”
In this approach, the FDA would expect a commitment from manufacturers and medical software developers on transparency and real-world performance monitoring for artificial intelligence and machine learning-based software as medical devices. In addition, periodic updates to the FDA on what changes and improvements are required as part of the approved pre-specifications and the algorithm change protocol.
Such a regulatory framework could enable the FDA and manufacturers to evaluate and monitor a medical software product or service from its premarket development to post-market performance and surveillance.
This approach could allow the FDA’s regulatory oversight to embrace the iterative improvement power of artificial intelligence and machine learning-based software as a medical device while assuring patient safety.
For SaMDs already registered in the Eu / FDA / Australia / Japan / Canada, it can be easily and quickly registered in Israel.
Good Machine Learning Practice
The FDA identified 10 guiding principles that can inform the development of Good Machine Learning Practice (GMLP).
These guiding principles will help promote safe, effective, and high-quality medical devices and software that use artificial intelligence and machine learning (AI/ML) technologies.
GMLP Guiding Principles
1. Multi-Disciplinary Expertise Is Leveraged Throughout the Total Product Life Cycle
An in-depth understanding of a model’s intended integration into clinical workflow and the desired benefits and associated patient risks can help ensure that ML-enabled medical devices are safe and effective and address clinically meaningful needs over the device’s lifecycle.
2. Good Software Engineering and Security Practices Are Implemented
Model design is implemented with attention to the “fundamentals”:
- Good software engineering practices
- Data quality assurance
- Data management
- Robust cybersecurity and patient privacy practices
These practices include a systematic risk management approach such as the one defined in ISO 14971. Moreover, the design process can appropriately capture and communicate design, implementation, and risk management decisions and rationale and ensure data authenticity and integrity.
3. Clinical Study Participants and Data Sets Are Representatives of the Intended Patient Population
Data collection and clinical evaluation protocols should be developed to ensure that the relevant characteristics of the intended patient population (for example, in terms of age, gender, sex, race, and ethnicity), use, and measurement inputs are sufficiently represented in a sample of adequate size in the clinical study and training and test datasets so that results can be reasonably generalized to the population of interest.
This is important to manage any bias, promote appropriate and generalizable performance across the intended patient population, assess usability, and identify circumstances where the model may underperform.
4. Training Data Sets Are Independent of Test Sets
Training and test datasets are selected and maintained to be appropriately independent of one another. All potential sources of dependence, including patient, data acquisition, and site factors, are considered and addressed to assure independence.
5. Selected Reference Datasets Are Based Upon the Best Available Methods
Accepted, the best available methods for developing a reference dataset (that is, a reference standard) ensure that clinically relevant, well-characterized, and high-quality data are collected and the limitations of the reference are understood. If available, accepted reference datasets in model development and testing that promote and demonstrate model robustness and generalizability across the intended patient population are used.
6. Model Design Is Tailored to the Available Data and Reflects the Intended Use of the Device
Model design is suited to the available data and supports the active mitigation of known and prioritized risks, like overfitting performance degradation, and security risks. The clinical benefits and risks related to the medical product/service are well understood and used to derive clinically meaningful performance goals for testing and support that the product can safely and effectively achieve its intended use. Considerations include the impact of both global and local performance and uncertainty/variability in the device inputs, outputs, intended patient populations, and clinical use conditions.
7. Focus Is Placed on the Performance of the Human-AI Team
Where the model has a “human in the loop,” human factors considerations and the human interpretability of the model outputs are addressed with emphasis on the performance of the Human-AI team, rather than just the performance of the model in isolation.
8. Testing Demonstrates Device Performance during Clinically Relevant Conditions
Statistically sound test plans are developed and executed to generate clinically relevant medical device or software performance information independently of the training data set.
Considerations include the following:
- Intended patient population
- Important subgroups
- Clinical environment and use by the Human-AI team
- Measurement inputs
- Potential confounding factors
9. Users Are Provided Clear, Essential Information
Users are provided ready access to clear, contextually relevant information that is appropriate for the intended audience (such as health care providers or patients) including:
- Medical device or software intended use
- Indications for use
- Performance of the model for appropriate subgroups
- Characteristics of the data used to train and test the model
- Acceptable inputs
- Known limitations
- User interface interpretation
- Clinical workflow integration of the model
Users are also aware of the medical software/device modifications, updates from real-world performance monitoring, the basis for decision-making when available, and a means to communicate product/service concerns to the developer.
10. Deployed Models Are Monitored for Performance and Re-training Risks are Managed
Deployed models have the capability to be monitored in “real world” use with a focus on maintained or improved safety and performance. Additionally, when models are periodically or continually trained after deployment, there are appropriate controls in place to manage risks of overfitting, unintended bias, or degradation of the model (for example, dataset drift) that may impact the safety and performance of the model as the Human-AI team uses it.
Quality Management System (QMS)
A medical device quality management system (QMS) such as ISO 13485 and/or FDA 21 CFR part 820 is a structured system of procedures and processes covering all aspects, such as:
- Design
- Manufacturing
- Supplier management
- Risk management
- Complaint handling
- Clinical data
- Storage
- Distribution
- Labeling
Most medical devices will require procedures, policies, protocols, and forms as part of the QMS. The complexity and implementation of the QMS will vary based on the classification of the device.
SaMD Risk Categorization
The IMDRF risk framework identifies the following two major factors as describing the intended use of the SaMD:
- Significance of information provided by the SaMD to the healthcare decision – Identifies the intended use of the information provided by the SaMD – i.e., to treat or diagnose; to drive clinical management; or to inform clinical management.
- State of healthcare situation or condition, which identifies the intended user, disease or condition, and the population for the SaMD – Defines the medical condition and vulnerability level of the patient that will use the medical device i.e., critical; serious; or non-serious healthcare situations or conditions.
Taken together, these factors describing the intended use can be used to place the AI/ML-based SaMD into one of four categories, from lowest (I) to highest risk (IV), to reflect the risk associated with the clinical situation and device use.
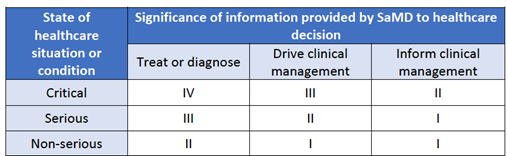
Table 1: FDA Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning-Based Software as a Medical Device
SaMD Locked & Adaptive algorithms
“Locked” algorithms that are part of the SaMD are those that provide the same result each time the same input is provided. As such, a locked algorithm applies a fixed function (e.g., a static look-up table, decision tree, or complex classifier) to a given set of inputs. These algorithms may use manual processes for updates and validation.
In contrast to a locked algorithm, an adaptive algorithm (e.g., a continuous learning algorithm) changes its behavior using a defined learning process.
The algorithm adaptation or changes are implemented such that for a given set of inputs, the output may differ before and after the changes are implemented.
These algorithm changes are typically implemented and validated through a well-defined and possibly fully automated process aiming to improve performance based on the analysis of new or additional data.
The adaptation process can be intended to address several different clinical aspects, such as:
- Optimizing performance within a specific environment (e.g., based on the local patient population)
- Optimizing performance based on how the medical software/device is being used (e.g., based on the preferences of a specific physician)
- Improving performance as more data are collected and/or changing the medical software/device’s intended use.
- New clinical data and clinical studies results
The adaptation process of medical software follows two stages- learning and updating:
| Adaption stage | Stage details |
| Learning | The algorithm “learns” how to change its behavior, for example, by adding new input types or new cases to an existing training database. |
| Updating | The “update” occurs when the algorithm’s new version is deployed. |
FDA 510K SaMD Modifications, Change Control and Regulation
There are many possible modifications, changes, and improvements to an AI/ML-based SaMD during the lifecycle of the medical software.
Some modifications and changes may not require a review based on guidance provided in “Deciding When to Submit a 510(k) for a Software Change to an Existing Device.”
This paper anticipates that many modifications to AI/ML-based SaMD involve algorithm architecture modifications and re-training with new data sets, which under the software modifications guidance would be subject to premarket review by the FDA, based on the risk involved with these modifications.
The types of SaMD modifications generally fall into three broad categories:
| Modification type | Modification details |
| Performance | Clinical and analytical performance |
| Inputs | Inputs used by the algorithm and their clinical association to the SaMD output |
| Intended use | The intended use of the SaMD, as outlined above and in the IMDRF risk categorization framework, is described through the significance of the information provided by the SaMD for the state of the healthcare situation or condition. |
The changes described may not be mutually exclusive – one medical software modification may impact, for example, both a change in input and change in performance; or, a performance change may increase a device’s/SaMD clinical performance that in turn impacts the intended use. These medical software changes in AI/MLbased SaMD, grouped by the types of changes described above, have a different impact on users, which may include either patients, healthcare professionals, or others
SaMD Validation and Verification
Verification and validation (V&V) for the medical software are checks and balances in the medical device/medical software design process that identify deficiencies and discrepancies in the design before the medical software/device is produced.
SaMD Verification
Verification is particularly important in medical software/ device design, while the validation is before the industrial manufacturing phase.
Verification ensures the output of each phase of the design process meets the requirements derived from the previous phase.
Verification activities may include worst-case analysis, fault tree analysis, inspection, testing, and failure mode and effects analysis.
Verification tests the output from each step against the previous step.
SaMD Validation
Software validation is defined as documented evidence with a high degree of assurance that the software functions as per software design and user requirements in a consistent and reproducible manner.
The validation ensures that the medical software/device meets the user’s needs and prioritizes risks.
In medical device design, verification and validation must be done to obtain approval for the device.
In simple words, the verification ensures the medical device/medical software conforms to the design criteria.
The validation ensures the medical device/medical software conforms to the user’s requirements
V&V can be especially complicated with AI.
As these machines “learn,” the pathways they take to arrive at decisions change, so the original programmers of the algorithms cannot tell how the decisions were arrived at.
This creates a “black box” that can be problematic for highly regulated industries such as pharma and medical, where the reasons for decisions and actions need to be clear, documented, and approved.
The FDA and other health authorities are considering questions posed by fluid and continual learning:
- Since a Continual Learning System (CLS) is dynamic, the algorithm changes over time. What are the criteria for updating the algorithm? Is it entirely automated? Or is there human involvement?
- Because the performance changes over time, can we monitor the performance in the field to better understand where the CLS is going?
- Understanding that users may be affected as the algorithm evolves and provides different responses, what is an effective way to communicate the changes?
- How do we ensure new data that leads to changes in the algorithm is of adequate quality?
SaMD Development Cycle
To fully realize the power of AI/ML learning algorithms while enabling continuous improvement of their performance and limiting degradations, the FDA’s proposed TPLC approach.
The TPLC approach for SaMD is based on the following general principles that balance the benefits and risks, and provide access to safe and effective AI/ML-based SaMD:
- Establish clear expectations, write and approve controlled documents accordingly on quality management systems (ISO 13485/21CFR part 820) and Good ML Practices (GMLP);
- Conduct premarket review for those SaMD that require premarket submission to demonstrate reasonable assurance of safety and effectiveness and establish clear expectations for manufacturers of AI/ML-based SaMD to manage patient risks throughout the lifecycle continually;
- Expect manufacturers to monitor the AI/ML device and incorporate risk management, change control, and other approaches outlined in “Deciding When to Submit a 510(k) for a Software Change to an Existing Device” in the development, validation, and execution of the algorithm changes (SaMD Pre-Specifications and Algorithm Change Protocol); and
- Enable increased transparency to users and FDA using postmarket real-world performance and post-marketing surveillance reporting to maintain continued safety and effectiveness assurance.
SaMD Pre-Specifications and Algorithm Change Protocols
SaMD Pre-Specifications (SPS)
A SaMD manufacturer’s anticipated modifications to performance, inputs, or changes related to the intended use of AI/ML-based SaMD.
The manufacturer plans to achieve these types of changes when the SaMD is in use.
The SPS draws a “region of potential changes” around the initial specifications and labeling of the original device. This is “what” the manufacturer intends the algorithm to become as it learns.
Algorithm Change Protocol (ACP)
Specific methods that a manufacturer has in place to achieve and appropriately control the risks of the anticipated types of modifications delineated in the SPS.
The ACP is a step-by-step delineation of the data and procedures to be followed so that the modification achieves its goals and the medical software/device remains safe and effective after the modification.
Learning, adaptation, and optimization are inherent to AI/ML-based SaMD
These capabilities of AI/ML would be considered modifications to SaMD after they have received market authorization from FDA.
This paper proposes an approach to appropriately manage patient risks from these modifications while enabling manufacturers to improve performance and potentially advance patient care.
In the medical software modifications guidance, depending on the type and the impact of change, manufacturers are expected to submit a new premarket submission if the modification is beyond the intended use for which the SaMD was previously authorized.
For this proposed approach, we anticipate that there may be cases where the SPS or ACP can be refined based on “the real-world” learning and training for the same intended use of the AI/ML SaMD model.
In those scenarios, FDA may conduct a “focused review” of the proposed SPS and ACP for a particular SaMD.
SaMD Real World Performance
Real-world performance monitoring may be achieved in various suggested mechanisms currently employed or under pilot at FDA, such as adding to file or an annual report, Case for Quality activities, or real-world performance analytics via the Pre-Cert Program.
Reporting type and frequency may be tailored based on the device’s risk, number, types of modifications, and maturity of the algorithm (i.e., quarterly reports are unlikely to be useful if the algorithm is at a mature stage with minimal changes in performance over the quarter).
Involvement in pilot programs, such as Case for Quality and the Pre-Cert Program, may also impact the reporting type and frequency, giving insight into the manufacturer’s TPLC and organization.
Participation in these programs could provide another avenue to support continued assurance of safety and effectiveness in the development and modifications of AI/ML-based SaMD.
SaMD FDA Regulation Drafts and Transparency
SaMD Manufacturers can work to assure the safety and effectiveness of their medical software products/services by implementing appropriate mechanisms that support transparency and real-world performance monitoring. Transparency about the function and modifications of medical devices is a key aspect of their safety.
This is especially important for medical devices, like SaMD that incorporate AI/ML, which change over time. Further, many of the modifications to AI/ML-based SaMD may be supported by the collection and monitoring of real-world data.
Gathering performance data on the real-world use of the SaMD may allow manufacturers to understand how their products/services are being used, identify opportunities for improvements, and respond proactively to safety or usability concerns.
Real-world data collection and monitoring is an important mechanism that manufacturers can leverage to mitigate the risk involved with AI/ML-based SaMD modifications, in support of the benefit-risk profile in the assessment of a particular AI/ML-based SaMD as well as for continuous improvement.
Through this framework, manufacturers would be expected to commit to the principles of transparency and real-world performance monitoring for AI/ML-based SaMD.
The FDA would also expect the manufacturer to provide periodic reporting to the FDA on updates that were implemented as part of the approved SPS and ACP, as well as performance metrics for those SaMD.
This commitment could be achieved through a variety of mechanisms.
Transparency may include updates to different bodies such as:
- FDA and other health authorities the medical software is registered in
- Medical software and device companies and collaborators of the manufacturer
- Clients, patients, and general users.
- Clinicians and hospitals
For modifications in the SPS and ACP, manufacturers would ensure that labeling changes accurately and completely describe the modification, including its rationale, any change in inputs, and the updated performance of the SaMD.
Manufacturers may also need to update the specifications or compatibility of any impacted supporting devices, accessories, or non-device components.
Finally, manufacturers may consider unique mechanisms for how to be transparent – they may wish to establish communication procedures that could describe how users will be notified of updates (e.g., letters, email, software notifications) and what information could be provided (e.g., how to appropriately describe performance changes between the current and previous version).
Artificial intelligence and machine learning will continue to evolve in healthcare settings and the E-Health industry.
To develop high-quality medical software, industry professionals should remain knowledgeable about up-to-date regulatory policy.
The FDA’s emphasizing transparency keeps developers, regulators, and patients confident that products were constructed with quality metrics and quality management systems.
Gain the advantage of bringing your SaMD to the global market by comprehending the developmental and regulatory journey.
This knowledge will empower you to secure approvals, complete registrations, and achieve international sales, including in regulated markets like the US, EU, Japan, Australia, Canada, and Israel.
Take the first step towards your success now!